

Beyond the Sequence: The Role of Structural Variants in Genomics
Structural variants (SVs) are having their moment in genomics, and it’s long overdue.
With their ability to reshape our understanding of genetic complexity, SVs hold the key to uncovering hidden genomic secrets. Yet, they’ve often taken a backseat to single-nucleotide variants (SNVs) and small insertions or deletions. Why? Largely due to past technological constraints and a practical research focus prioritized on detecting simpler, more easily measurable genomic changes.
However, this is changing. Advances in genome mapping and sequencing technologies have not only made SV detection and analysis easier but have also positioned them as central to modern genomic research. Whether you’re deep into molecular biology, invested in cytogenetics, or simply curious, understanding SVs is critical for anyone in the field. Here’s why SVs matter, and how integrating molecular and cytogenetic perspectives can take genomic insights to a whole new level.
The Role of SVs in Genomics
What are SVs?
SVs are large alterations in the genome, ranging in size from 50 to millions of base pairs. Unlike the more commonly discussed SNVs that change a single base pair, SVs include a range of structural rearrangements such as:
- Insertions: The addition of new genetic material into a DNA sequence.
- Deletions: The removal or loss of genetic material from a DNA sequence.
- Duplications: The replication of entire genes or genomic regions, resulting in additional copies of the segment.
- Inversions: A reversal of the orientation of a segment of DNA within the sequence.
- Translocations: The relocation of DNA segments to different positions, either within the same chromosome or to a different chromosome.
Many SVs are not rare; they make up a significant portion of genomic variation in humans. They influence gene regulation, protein function, and chromosomal structure, making them key players in both normal biological processes and disease.
Why SVs Outweigh SNVs in Complexity and Impact
While SNVs explain many Mendelian disorders, SVs are often responsible for more complex genetic phenotypes and diseases—the ones that keep clinicians and families up at night. SVs are the variants behind some of the most devastating diagnoses:
- Neurodevelopmental Disorders: Large copy-number variants have been implicated in conditions like autism and developmental delays, affecting up to 15% of children with one of these diagnoses.1
- Cancer: Chromosomal rearrangements, such as the famous Philadelphia chromosome (BCR-ABL1 fusion), are hallmark drivers of various cancers, including chronic myelogenous leukemia (CML).2 In pediatric oncology, high-impact SVs are often the culprits behind aggressive, treatment-resistant tumors.
- Rare Diseases: SVs frequently go undetected but play a critical role in many rare diseases, like 22q11.2 deletion syndrome.3 For many families, identifying the underlying SV is the first step in ending a long diagnostic odyssey.
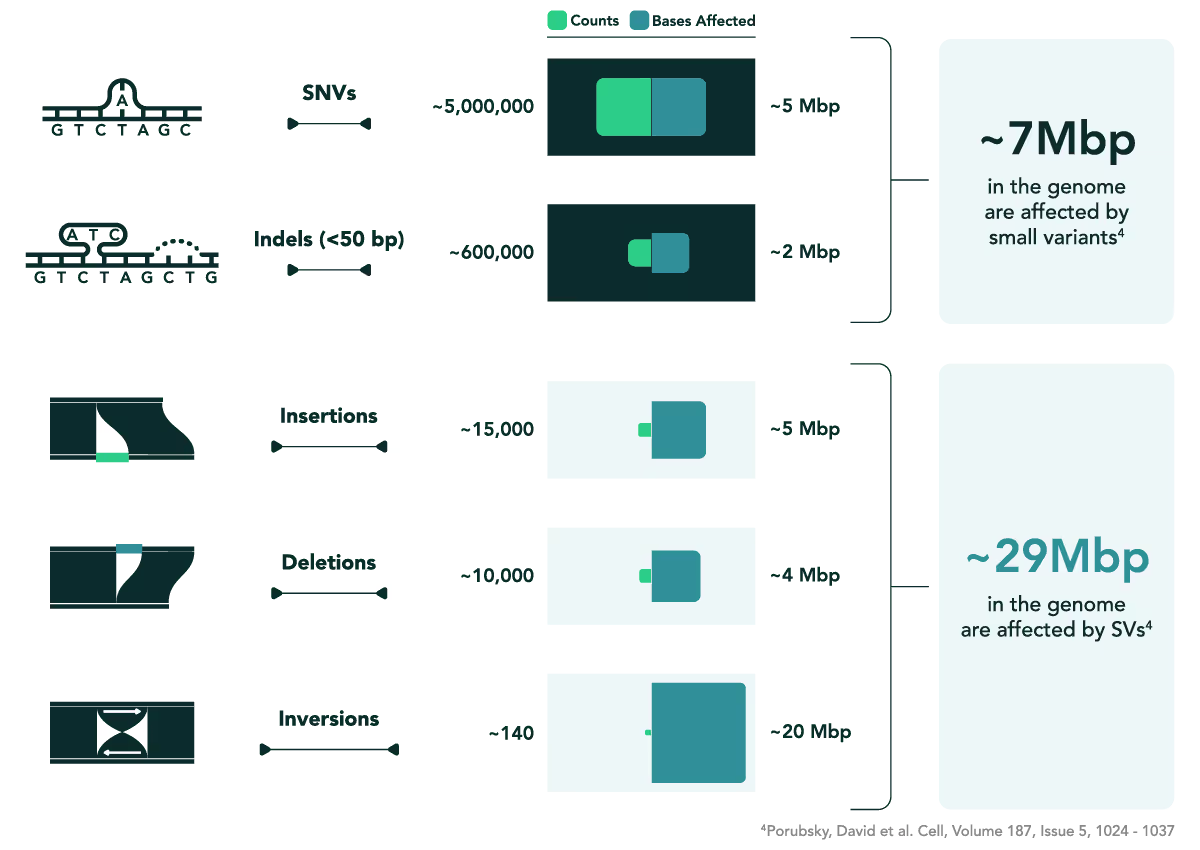
While SNVs are often abundant across the genome, their individual effects tend to be subtler compared to the sweeping changes brought about by SVs.4 Because of their size and complexity, SVs often affect multiple genes or regulatory regions, amplifying their power to alter phenotypes.
To truly understand—and one day treat or prevent—the conditions that disrupt lives, challenge hope, and rewrite futures, we need to place SVs front and center in our study of the human genome.
Rethinking SV Analysis
For decades, SVs have been studied through the separate lenses of molecular biology and cytogenetics. Each approach has its strengths, but each also has limitations that, until recently, left a gap in our understanding.
Molecular Methods
Molecular tools like next-generation sequencing (NGS) have driven incredible progress in detecting SVs at the nucleotide level. These technologies excel in identifying precise breakpoints and characterizing the functional consequences of SVs. For instance, NGS has revolutionized our ability to characterize translocations like the BCR-ABL1 fusion gene in CML, providing precise molecular details about breakpoints and functional consequences.5 This fusion, where a portion of chromosome 9 swaps places with part of chromosome 22, not only enhanced diagnostic precision but also highlighted an opportunity for targeted treatment. The ability to pinpoint such SVs at the molecular level has provided lifesaving therapeutic insights.
Despite these advances, the cost considerations vary significantly—while short-read sequencing has become highly accessible, long-read sequencing technologies that excel at detecting longer SVs remain considerably more expensive. Further, molecular methods often operate from a zoomed-in perspective, focusing on granular details at the expense of genome-wide context. While they reveal small-scale effects with incredible clarity, this microscopic focus can make it difficult to situate these variations within the bigger picture of genomic architecture.
Cytogenetic Methods
On the other end of the spectrum, cytogenetics has historically excelled at capturing the broader genomic landscape. Techniques like karyotyping and fluorescence in situ hybridization (FISH) have been foundational in visualizing how chromosomes are structurally organized and how they change. A classic example is the detection of the 22q11.2 deletion syndrome, where a small missing piece of chromosome 22 results in a complex array of symptoms, from congenital heart defects to learning challenges.4 Cytogenetics revealed this deletion as a hallmark of the syndrome, providing a critical diagnostic tool for clinicians tackling diverse patient presentations.
However, while cytogenetic methods can uncover different types of SVs like deletions and translocations, their resolution often limits investigations to larger-scale rearrangements. This lack of fine detail can leave researchers with an incomplete view of how SVs exert their influence at the molecular level. In addition, cytogenetic approaches can be costly per sample, with expenses varying significantly depending on the specific method and scope of analysis required.
Bridging the Gap with Electronic Genome Mapping
For decades, advancing our understanding of SVs meant leveraging the strengths of molecular and cytogenetic approaches separately, despite their limitations. But today, technologies like Electronic Genome Mapping (EGM) are bridging the gap between high-resolution molecular analysis and genome-wide structural assessment. EGM unites the detailed insights of molecular methods with the expansive coverage of cytogenetics, offering a hybrid approach that captures both small-scale intricacies and the wider context of genomic rearrangements. EGM's economic advantage may lie in consolidating multiple tests into a single comprehensive analysis, potentially reducing the cumulative costs of sequential cytogenetic and molecular approaches.
EGM doesn’t just fill gaps; it creates new opportunities for discovery by uniting previously isolated approaches, equipping scientists to see the genome with unparalleled clarity.
The Future of SV Research
SVs represent some of the most powerful drivers of genomic complexity, evolution, and disease. By understanding their roles—as evidenced in diseases like CML or 22q11.2 deletion syndrome—scientists can not only diagnose conditions more accurately but also create targeted, effective treatments.
Tools like EGM are helping to transform how we study and leverage SV data, moving the field toward a fully integrated approach. With molecular and cytogenetic methods working harmoniously, the future of SV research looks brighter than ever.
By putting SVs at the forefront, we’re not just uncovering the mechanics of the genome—we’re unlocking the next frontier in biology. Together, as a community of researchers and innovators, we hold the tools to redefine what’s possible in genomics, one SV at a time.
Citations
- Cooper, G. M. et al. A copy number variation morbidity map of developmental delay. Nat. Genet. 43, 838–846 (2011).
- Rudkin, G. T., Hungerford, D. A. & Nowell, P. C. DNA Contents of Chromosome Ph1 and Chromosome 21 in Human Chronic Granulocytic Leukemia. Science 144, 1229–1232 (1964).
- Kelley, R. I. et al. The association of the DiGeorge anomalad with partial monosomy of chromosome 22. J. Pediatr. 101, 197–200 (1982).
- Porubsky, D. & Eichler, E. E. A 25-year odyssey of genomic technology advances and structural variant discovery. Cell 187, 1024–1037 (2024).
- Linhartova, J. et al. Characterization of 46 patient-specific BCR-ABL1 fusions and detection of SNPs upstream and downstream the breakpoints in chronic myeloid leukemia using next generation sequencing. Mol. Cancer 14, 89 (2015).
