

Why Detection Physics Matters for Genome Mapping
Introduction
How we detect DNA matters just as much as how we analyze it.
Over the past decade, improvements in mapping high molecular weight (HMW) DNA have expanded what researchers can see across the genome, especially for structural variation and repeat-rich regions. As applications push toward denser labeling and finer-scale features, the limits of detection itself have become harder to ignore.
At its core, genome mapping is a detection problem. Some platforms rely on light, using fluorescence and imaging systems to visualize labeled DNA molecules. Others operate entirely in the electrical domain. These approaches differ not only in instrumentation, but also in the physical signals they measure—and in the limits those signals impose on resolution, scalability, and cost.
Most comparisons between optical genome mapping (OGM) and electronic genome mapping (EGM) focus on downstream performance metrics. Here, the emphasis is upstream instead—on the physics and workflow architecture that shape those outcomes.
Optical Genome Mapping
Detection Physics
.webp)
OGM translates molecular features into spatial information by imaging light emitted from fluorescent labels attached to HMW DNA. In a typical workflow, labeled DNA molecules are elongated—often within nanochannels or on a surface—illuminated with a laser, and visualized through a microscope objective.1 Each fluorescent tag emits photons that are collected, focused, and projected onto a detector, producing a two-dimensional image of label positions along the molecule.
The key assumption in this approach is that spatial separation along the DNA can be faithfully converted into spatial separation in the image. In practice, that conversion is limited by the physics of light itself.
Light behaves as a wave, and when it passes through an optical system, it cannot be focused to an infinitesimally small point. Even a single point source produces a finite intensity distribution on the detector. When two fluorescent labels are far enough apart, their intensity distributions remain distinct, but as they move closer together, those distributions begin to overlap.
Below a certain separation distance, the system can no longer reliably distinguish the signals as two independent features. This diffraction limit sets a fundamental constraint on optical resolution. For visible wavelengths and realistic numerical apertures, this limit is typically about 200–250 nanometers. Importantly, this distance is measured in physical space, not in base pairs along the DNA. When labeled motifs are closer than this distance after elongation, their optical signatures merge, regardless of labeling accuracy or downstream analysis.
For genome mapping, this has direct consequences. As labeling density increases—either because sequence motifs occur frequently in certain regions or because higher-resolution maps are desired—distinct molecular features can collapse into a single optical signal. Once this overlap occurs, the original spacing information is no longer present in the image, and no amount of image processing or computational refinement can recover separations that were never optically resolved.
Efforts to push optical resolution further tend to run into steep tradeoffs. Higher numerical aperture objectives improve resolving power but reduce depth of field and increase sensitivity to mechanical drift. Brighter or more photostable fluorophores can improve signal-to-noise but bring added constraints around labeling chemistry and photobleaching. Super-resolution methods can bypass diffraction in some imaging contexts, but they add substantial complexity, longer acquisition times, and experimental requirements that are hard to reconcile with high-throughput genome mapping.
These physical limits also shape how optical systems scale. Because detection relies on imaging extended physical areas, increasing throughput generally means scanning larger regions, imaging faster, or running longer experiments. Each approach amplifies demands on optics, motion control, illumination stability, and data handling. As a result, improvements in resolution and throughput are tightly coupled to increases in system complexity and cost.
Cost Drivers
Because optical genome mapping relies on imaging fluorescent labels, the physical limits described above directly shape how these systems scale and where costs accumulate. Detecting fluorescent labels requires a stack of precision components—lasers, objectives, cameras, illumination optics, and motion control hardware. Each of these elements must operate within narrow tolerances to maintain focus, alignment, and signal quality across large imaging areas.
Throughput in optical systems is inherently area- and time-bound. To map more molecules, the system must image more physical space, image faster, or both. Imaging larger areas pushes toward bigger fields of view, higher-end objectives, and larger or more sensitive cameras. Imaging faster places additional demands on illumination stability, camera performance, and mechanical precision. In either case, gains in throughput usually come with increases in system complexity and cost.
Resolution follows a similar pattern. Approaching the diffraction limit requires higher numerical aperture optics, improved signal-to-noise, and tighter mechanical control. These improvements raise both the cost and fragility of the system, while still leaving the fundamental resolution ceiling in place. As a result, optical platforms often face tradeoffs between resolution, throughput, and cost that reflect the physics of light rather than software or workflow choices.
Consumables and run time also factor into cost. Fluorophores, photostable dyes, and extended imaging times contribute to per-sample expense, particularly as experiments move toward higher labeling density or longer molecules. Taken together, these elements mean that cost in optical genome mapping scales closely with imaging demands.
Electronic Genome Mapping
Detection Physics
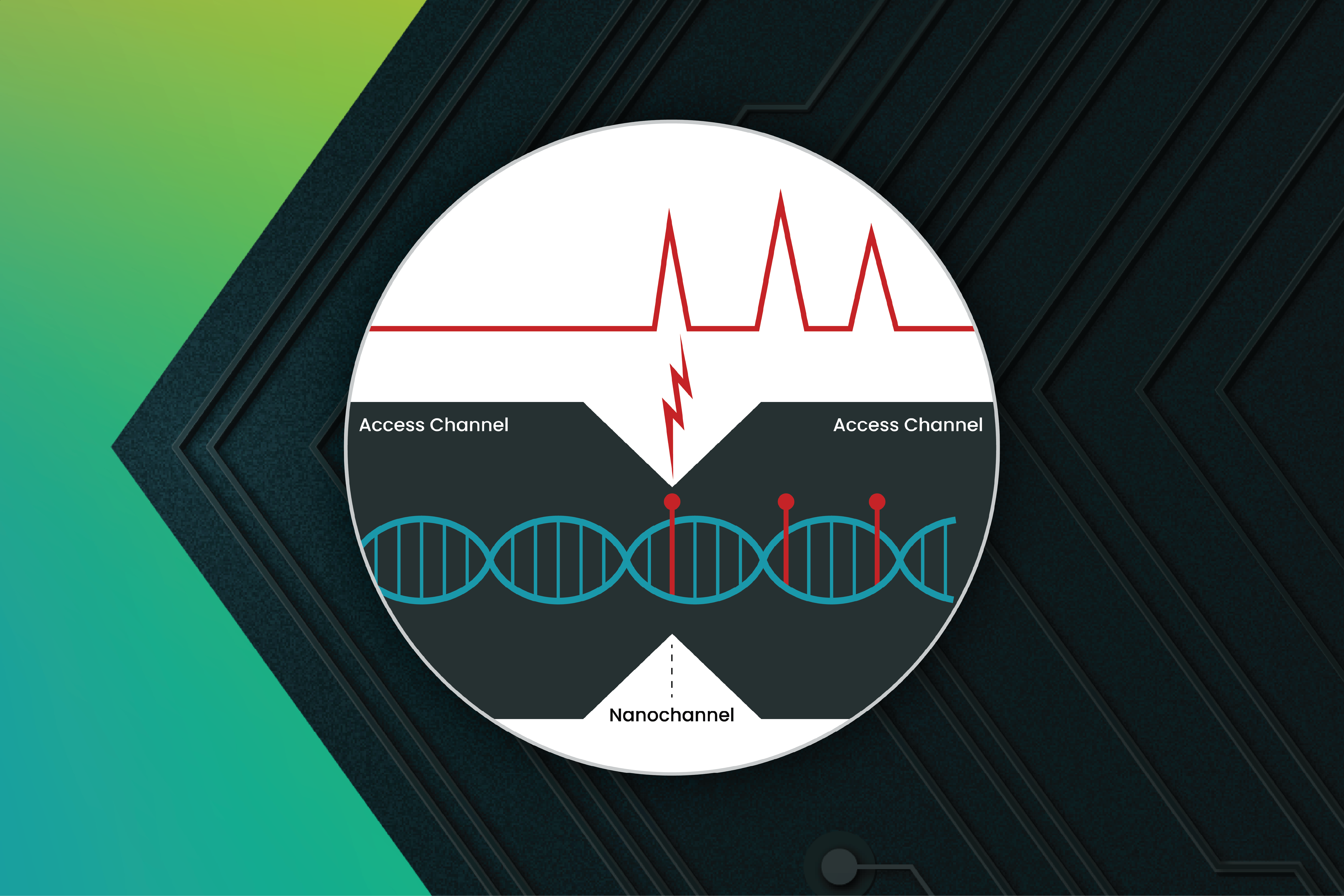
Electronic genome mapping approaches the same problem—detecting labeled DNA—from a fundamentally different physical starting point. Rather than converting molecular features into images, it operates entirely in the electrical domain, measuring how molecules perturb ionic flow as they pass through nanodetectors. Instead of forming an image, the system measures changes in the voltage across paired sensors as HMW DNA molecules move through a confined nanochannel.2
The applied electric field plays two roles. First, it is required for electronic sensing, since the baseline signal arises from ions carrying current through electrolyte-filled channels. Second, it provides the electrophoretic force that drives molecules into the nanochannel and through it.
EGM operates as a current clamp, where the measured voltage is determined by the resistance of the fluidic path. A useful way to think about the detector is as a simple series circuit made up of three resistive elements: an access channel to the left of the nanochannel, the nanochannel sensing region itself, and an access channel to the right of the nanochannel. When RecA-coated DNA enters the sensing region, it displaces ions and increases the effective resistance locally. With the current held constant, that resistance change appears as a measurable change in the voltage across paired sensors positioned close to the constriction.
Tagged sites are detected through the same physical mechanism. As a tag passes through the nanochannel, its additional bulk displaces more ions than the RecA-coated backbone alone, producing a distinct voltage feature that can be detected and timed. Tagged sites generate characteristic electrical signatures that are separable from the backbone signal and from background fluctuations.
This electronics-based sensing framework also changes what limits resolution. EGM does not rely on spatial imaging, so it does not face diffraction-limited overlap of neighboring features. Because detection occurs entirely in the electrical domain, there are no lenses, cameras, or fluorescent signals setting the resolution limit. Instead, separability depends on the geometry of the sensing region and the temporal precision of the voltage measurement. Closely spaced tagged sites can remain distinct as long as they pass through the detector sequentially, producing resolvable voltage features in time.
This time-based separability introduces its own tradeoffs. As molecule velocity increases, the temporal spacing between features decreases, making it more challenging to resolve closely spaced events. In practice, achieving higher resolution may require operating at lower translocation speeds, which can reduce per-detector throughput and create a balance between resolution and processing rate.
Distance along the DNA molecule is reconstructed from the timing between successive tag-associated voltage changes, converting temporal information into physical spacing. This time-based measurement, combined with a confined sensing region, preserves information density in regions where imaging-based detection begins to lose detail.
Cost Drivers
In EGM, cost is driven by detector architecture and electronics rather than imaging. Because detection occurs entirely in the electrical domain, there is no optical stack to scale—no lenses to enlarge, no fields of view to expand, and no cameras whose size or sensitivity must increase as throughput grows.
Throughput scales by adding detectors in parallel. Each nanochannel detector operates independently, producing its own voltage trace. Increasing throughput, therefore, means increasing detector count and improving electronic readout, not expanding physical imaging area. This parallelism allows performance gains to come from denser integration rather than from larger or more complex mechanical assemblies.
The electronics that support detection follow a different cost curve than optical components. Amplifiers, analog-to-digital converters, and on-chip signal routing benefit from decades of work and optimization in the semiconductor industry. As detector density increases, additional nanodetectors can be integrated at relatively modest cost, especially as designs move toward more on-chip electronics and shared readout architectures.
Importantly, resolution improvements do not require more expensive hardware in the same way they do for optical systems. Because separability is set by sensing geometry and temporal precision rather than by wavelength, refining detector design or signal processing can increase information density without introducing fundamentally new classes of components. This decoupling of resolution from hardware scale is a major reason EGM follows a different cost trajectory.
Detection Shapes the Difference
OGM and EGM are shaped by fundamentally different detection physics, which in turn define how resolution, throughput, and cost scale. Optical systems are constrained by the physics of imaging, while electronic systems operate through voltage measurements governed by charge and resistance instead.
As applications demand higher information density, tighter resolution, and scalable throughput, those physical foundations matter. Imaging-based systems must contend with diffraction, expanding fields of view, and growing optical complexity. Electronic systems follow a different path, one where resolution is set by detection geometry and timing, and where throughput scales with parallel detectors and electronics rather than larger microscopes.
For researchers working with HMW DNA, structural variation, or repeat-rich regions, understanding these detection differences helps clarify where EGM fits. It doesn’t replace sequencing or existing mapping approaches, but it offers a fundamentally different way to extract long-range genomic structure with high resolution and a cost profile that reflects its electronic rather than optical roots.
Citations
1. Jeffet J, Margalit S, Michaeli Y, Ebenstein Y. Single-molecule optical genome mapping in nanochannels: multidisciplinarity at the nanoscale. Essays Biochem. 2021;65(1):51-66. doi:10.1042/EBC20200021
2. Oliver JS, Catalano A, Davis JR, et al. High-Definition Electronic Genome Maps from Single Molecule Data. bioRxiv. Preprint posted online May 18, 2017:139840. doi:10.1101/139840
