

What Structural Variants May Reveal About Unsolved Rare Disease Cases
The Persistent Diagnostic Gap in Rare Disease
Over the past two decades, advances in genomic sequencing have transformed rare disease research and diagnostics. Today, sequencing-based workflows can resolve the genetic basis of disease in approximately 30–50% of cases, with higher yields reported in certain cohorts and clinical contexts.1,2 Yet for a substantial fraction of patients, even extensive testing fails to yield a definitive result.
This unresolved 50–70% represents one of the most persistent challenges in rare disease genomics. Despite whole-genome sequencing, reanalysis pipelines, and expanding variant databases, many cases remain unexplained. As the field reflects on more than 25 years since the first human genome was sequenced, a fundamental question remains—what types of genomic variation may still be missing from standard approaches?
Increasingly, attention is turning toward genome structure and the possibility that structural variants (SVs) may play an underappreciated role in the unsolved fraction of rare disease.
What We Mean by Structural Variants
SVs are genomic changes larger than 50 base pairs that alter the organization of DNA rather than individual nucleotides. These variants include insertions, deletions, duplications, inversions, translocations, and more complex rearrangements that can span kilobases to megabases of sequence. For more information on SVs, check out Beyond the Sequence: The Role of Structural Variants in Genomics.
Why Rare Disease Remains Partially Unsolved
The Limits of a Sequencing-Centered View
Much of modern rare disease genomics has been shaped by the strengths of short-read sequencing (SRS). These technologies excel at detecting single-nucleotide variants (SNVs) and small insertions or deletions, enabling large-scale studies with remarkable throughput and affordability. As sequencing costs declined, whole-genome analysis became increasingly routine across both research and clinical settings.
However, this success has also narrowed the scope of variation most commonly interrogated. SRS is optimized for small-scale changes, and many analytical pipelines are built around the interpretation of SNVs. Variants that depend on long-range genomic context—such as large rearrangements, repeat-associated changes, or balanced events—remain difficult to detect or interpret using sequencing alone.
Long-read sequencing has expanded the range of detectable SVs, particularly for repeat expansions and larger insertions and deletions. However, even with single-molecule approaches, resolving highly complex or genome-wide rearrangements can be challenging, as these events often require broader structural context across entire chromosomes.
While small variants are far more numerous, larger structural changes often span a substantially greater portion of the genome.3 As a result, the types of questions researchers can ask are often constrained by the types of variation their tools can readily capture—with small, easily counted variants receiving disproportionate attention relative to larger, long-range SVs.
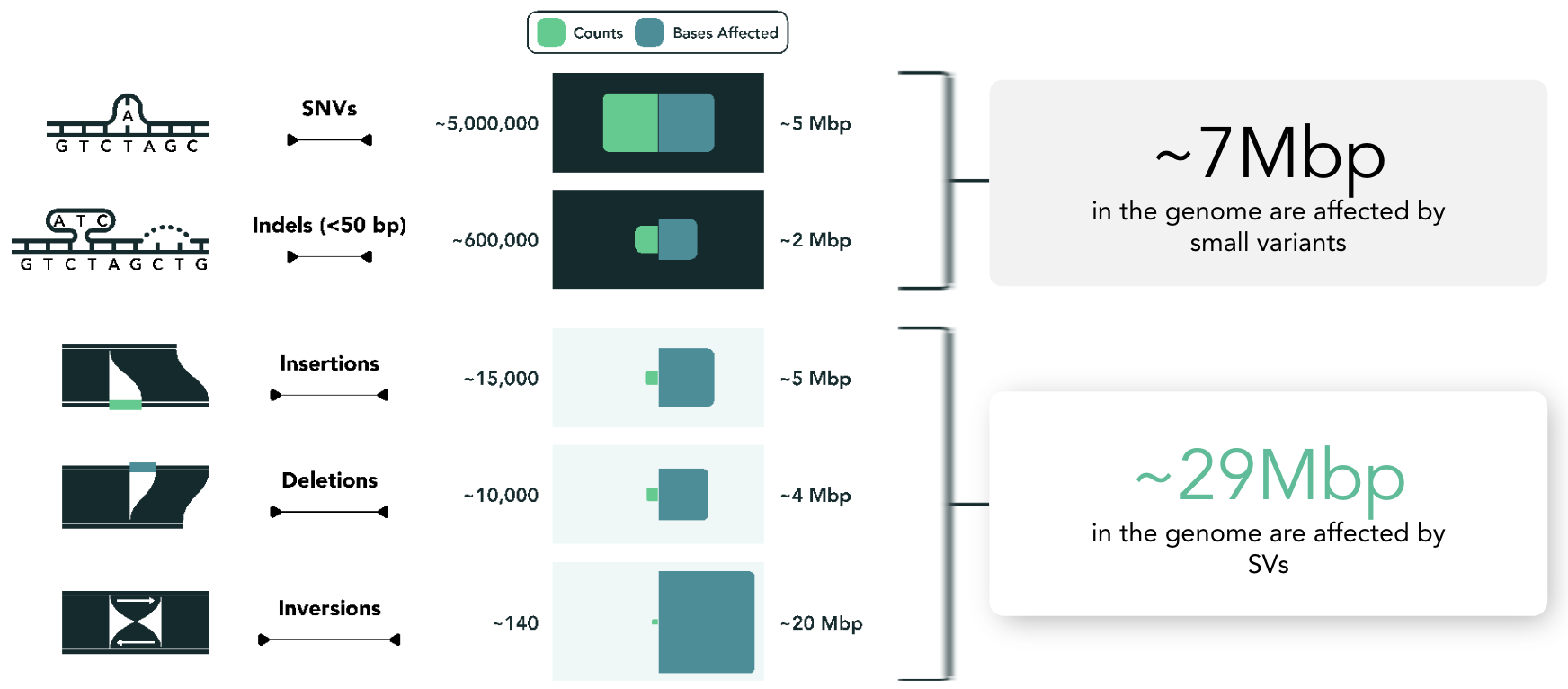
Structural and Interpretive Blind Spots
Beyond detection, interpretation presents an additional challenge. Because SV detection in SRS datasets relies heavily on computational inference, results can be difficult to interpret and may not always resolve variants unambiguously. Even when SVs are identified, their functional impact can be difficult to assess. Many SVs do not disrupt coding regions directly, but instead alter regulatory elements, chromatin organization, or long-range gene interactions. These effects are not always well represented in existing annotation frameworks.
Repetitive regions, balanced rearrangements, and complex de novo events are particularly challenging. In rare disease cases where sequencing yields inconclusive results, these blind spots raise the possibility that disease-relevant variation may be present—but unresolved—within the genome.
Emerging Evidence Linking Structural Variants to Rare Disease
Recent studies—and early findings emerging from rare disease cohorts—are beginning to explore this possibility in more detail. In a 2025 Nature Communications study, Jung and colleagues examined the role of complex de novo SVs in rare disorders, suggesting that such variants may be under-recognized contributors to pathogenic variation.4
Rather than identifying a single class of disease-causing SVs, the study highlights a broader methodological insight: when long-range genomic context is preserved, complex rearrangements can be reconstructed more accurately. Some variants that appear fragmented or ambiguous in sequencing-only data emerge as coherent events when structural information is incorporated.
Importantly, this work does not quantify how much of the diagnostic gap SVs may explain. Instead, it underscores a growing realization within the field: current workflows do not yet capture the full spectrum of disease-relevant genomic variation, particularly when it comes to complex structural changes. Though the available evidence is limited, these patterns are not confined to the literature alone.
In parallel, researchers working directly with rare and undiagnosed cohorts are applying structural-variant–focused analyses to revisit cases that remain unresolved after standard testing. In a sponsored research presentation, Catherine Brownstein described work at Boston Children’s Hospital and the Manton Center for Orphan Disease Research, which includes roughly 4,000 affected individuals from 3,800 families, spanning all 50 U.S. states and 71 countries. She emphasized that many of these cases remain undiagnosed even after exome or genome sequencing and highlighted examples in which revisiting long-standing cases with additional structural context has surfaced new, testable hypotheses.
In one such case, a suspected structural variant in the PHEX gene associated with X-linked hypophosphatemia was initially identified through sequencing and optical genome mapping, but its precise structure and interpretation remained uncertain. Using electronic genome mapping, researchers verified a ~60 kb tandem duplication and refined its genomic context, providing orthogonal confirmation and supporting downstream efforts to determine its relevance to disease. Check out the Boston Children’s Hospital Case Study to learn more.
While individual cases require careful follow-up and independent validation, these examples illustrate how incorporating structural context can help clarify ambiguous findings and generate more actionable hypotheses in rare disease research.
Do Structural Variants Explain the Remaining 50–70%?
At present, this remains an open question. There is no evidence to suggest that SVs alone account for the entirety—or even the majority—of unresolved rare disease cases. Assigning percentages without comprehensive data would be premature.
What is becoming increasingly clear, however, is that a predominantly SNV-focused view of the genome provides only a partial picture. Structural variation represents a plausible and historically underexplored contributor to missing heritability, particularly in cases where extensive sequencing has failed to yield clear answers, with many candidate findings still requiring careful interpretation and follow-up.
As genomics moves toward higher-quality diploid genome assemblies, interest is growing in hybrid analytical strategies that combine the throughput of SRS with technologies capable of preserving long-range structural context across entire molecules or chromosomes. These approaches aim not to replace sequencing, but to complement it—using long molecules as maps that provide the structural framework needed to interpret sequence-level variation more fully.
To read more about technologies suited to studying longer-range, structural genomic changes, check out Mapping the Shape of the Genomic World.
Implications for Rare Disease Genomics
For rare disease research and diagnostics, this shift has important implications. Addressing unresolved cases may require broadening the definition of actionable variation and integrating multiple data types into interpretation workflows.
Rather than relying on a single modality, future approaches are likely to emphasize complementarity—pairing sequence information with structural insight to better understand genome organization, variant phase, and complex rearrangements. Such integration may improve hypothesis generation, reduce interpretive uncertainty, and ultimately increase confidence in variant classification.
Crucially, this perspective reframes the diagnostic gap not as a failure of sequencing, but as a signal that additional dimensions of the genome remain to be explored.
Broadening the Variant Landscape
Rare disease genomics has achieved extraordinary progress, yet important questions remain unanswered. SVs may represent one piece of this unresolved puzzle, highlighting the need to expand how genomic variation is defined, detected, and interpreted.
As research continues to investigate the full architecture of the human genome, incorporating structural variation into rare disease analysis offers an opportunity to move beyond incremental gains toward deeper understanding. Closing the remaining diagnostic gap will likely depend not on any single technology, but on a more complete view of the genome itself.
Citations
1. Albuquerque ALB, dos Santos GG, Sadok SH, et al. Diagnostic Yield of Genome Sequencing Versus Exome Sequencing in Pediatric Patients With Rare Phenotypes: A Systematic Review and Meta-Analysis. Am J Med Genet A. 2025;197(10):e64146. doi:10.1002/ajmg.a.64146
2. Pandey R, Brennan NF, Trachana K, et al. A meta-analysis of diagnostic yield and clinical utility of genome and exome sequencing in pediatric rare and undiagnosed genetic diseases. Genet Med Off J Am Coll Med Genet. 2025;27(6):101398. doi:10.1016/j.gim.2025.101398
3. Porubsky D, Eichler EE. A 25-year odyssey of genomic technology advances and structural variant discovery. Cell. 2024;187(5):1024-1037. doi:10.1016/j.cell.2024.01.002
4. Jung H, Yang TP, Walker S, et al. Complex de novo structural variants are an underestimated cause of rare disorders. Nat Commun. 2025;16(1):9528. doi:10.1038/s41467-025-64722-2
